|
WYDZIAŁ
CHEMII - ZAKŁAD DYDAKTYCZNY TECHNOLOGII
CHEMICZNEJ
DESTYLACJA PODSTAWY TEORETYCZNE (Materiał pomocniczy do Ćwiczeń nr 4 i 12 oraz 15 i 22)
WIADOMOŚCI OGÓLNE
Destylacja jest operacją jednostkową, której celem jest rozdzielenie mieszaniny
cieczy na jej składniki. Polega ona na odparowaniu cieczy, odebraniu i
skropleniu powstałych par. Ciecz skroplona bogatsza w składnik lotniejszy
nazywana jest destylatem, natomiast pozostałość nieodparowana nazywa się
cieczą wyczerpaną (pogonem). Podstawę rozdziału stanowi tutaj różnica w
lotności składników. Efektywność rozdzielania na drodze destylacji jest
w wielu przypadkach niewystarczająca. Aby otrzymać czyste składniki konieczne
jest wtedy wielokrotne odparowanie cieczy i skroplenie par.
Destylacja i jej specyficzna odmiana - rektyfikacja są dyfuzyjnymi operacjami jednostkowymi. Pomimo, że są energochłonne znajdują szerokie zastosowanie - w laboratoriach, a w szczególności w przemyśle chemicznym, fermentacyjnym i farmaceutycznym. Pozwalają one na rozdzielanie ciekłych surowców naturalnych, mieszanin poreakcyjnych dwu - i wieloskładnikowych. Otrzymywane produkty charakteryzują się wysokim stopniem czystości. Wielokrotne
odparowanie cieczy i skroplenie par można uzyskać albo powtarzając wielokrotnie
destylację lub przeprowadzając jedną operację nazywaną rektyfikacją.
Ogólnie
mieszaniny z jedną fazą ciekłą w temperaturze wrzenia nazywa się homogenicznymi,
a w przypadku występowania w tej temperaturze kilku faz ciekłych heterogenicznymi.
Mieszaniny ciekłe dwuskładnikowe (binarne) dzielimy na: -
mieszaniny
idealne (doskonałe), w których siły oddziaływań międzycząsteczkowych
pomiędzy cząsteczkami wszystkich składników obecnych w układzie są zawsze
takie same oraz mieszaniny rzeczywiste.
Mieszaniny idealne spełniają prawo Raoulta., zgodnie z którym:
Ciśnienie cząstkowe składnika mieszaniny ciekłej nad mieszaniną jest wprost proporcjonalna do ułamka molowego tego składnika w mieszaninie co
wyraża się wzorem
pi = P0i x gdzie: pi - ciśnienia cząstkowe par składnika (i) nad mieszaniną; P0i - ciśnienie par składnika (i)nad czystym składnikiem (i); x- ułamek molowy składnika (i) w fazie ciekłej.
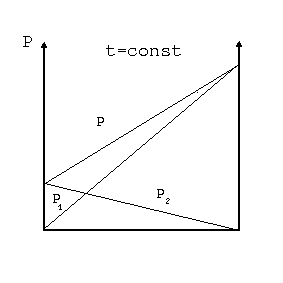
Ciśnienie całkowite P pary nad mieszaniną (w stalej temoaraturze) jest więc równe sumie ciśnień cząstkowych składników: dla mieszaniny dwuskładnikowej (binarnej) - patrz rys.1:
rys.1
Mieszaniny rzeczywiste dzielą się na - mieszaniny, w których siły przyciągania między cząsteczkami różnych składników są większe niż siły przyciągania między cząsteczkami tego samego typu. Występują wówczas ujemne odchylenia od prawa Raoulta. - mieszaniny, w których siły przyciągania między cząsteczkami różnych składników są mniejsze od oddziaływań między cząsteczkami tego samego rodzaju. W tych przypadkach mówimy o dodatnich odchyleniach od prawa Raoulta.
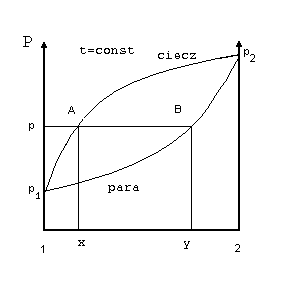
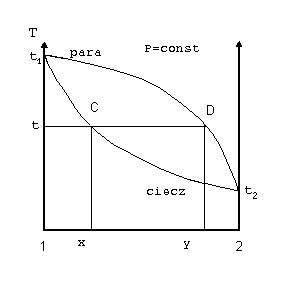
Rozdzielenie mieszanin ciekłych poprzez destylację i rektyfikację jest w głównej mierze uwarunkowane charakterem krzywych cieczy i pary przedstawiających wykres równowagi ciecz-para. Przebieg tych krzywych stanowi podstawę następującej klasyfikacji mieszanin cieczy o nieograniczonej wzajemnej mieszalności: Dla układu binarnego krzywe równowagi ciecz- para: - izoterma (stała temperatura) i izobara (stałe ciśnienie), w przypadku ogólnym; wyglądają następująco:
gdzie: 1, 2 – składniki „1” i „2” mieszaniny binarnej; X - ułamek molowy składnika lotniejszego w cieczy; Y - ułamek molowy składnika lotniejszego w parze; p1, p2 – prężność par nad czystym składnikiem; t1, t2 – temperatury wrzenia czystych składników; pkt. A, B i C, D odpowiadają składom fazy cieklej i gazowej w stanie równowagi ciecz-para.
Jak widać z powyższych rysunków skład pary różni się od składu cieczy większą zawartością składnika lotniejszego (wrzącego w niższej temperaturze).
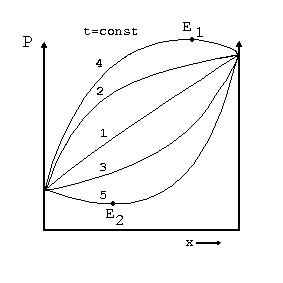
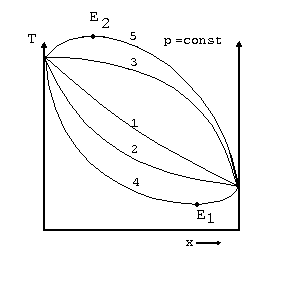
Rozdzielenie mieszanin ciekłych poprzez destylację i rektyfikację jest w głównej mierze uwarunkowane Bardziej szczegółowe rozważenie przebiegu krzywych cieczy i pary na wykresie równowagi ciecz-para. wskazuje na występowanie dwóch przypadków: równowag z punktem ekstremalnym (wykresy 4 i 5 na rys. 4 i 5) oraz równowag bez punktu ekstremalnego. Mieszaniny, dla których na krzywej P=f(x) oraz T=f(x), nie występują punkty ekstremalne nazywa się zeotropami. Mieszaniny wykazujące punkty ekstremalne na tych krzywych to mieszaniny azeotropowe - patrz rys.4 i rys.5.
Wykres równowagi (P,x i T,x) dla mieszanin: 1 - doskonałej, 2 - zeotropowej dodatniej, 3 - zeotropowej ujemnej, 4 -azeotropowej dodatniej, 5 - mieszaniny azeotropowej ujemnej, E - azeotropowe punkty ekstremalne.
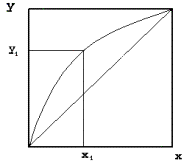
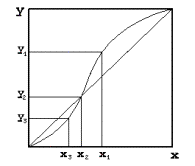
Równowagę ciecz-para wygodnie jest analizować we współrzędnych x-y. Korzysta się wówczas z tzw. kwadratu jednostkowego odkładając na osiach ułamki molowe (stężenie) składnika bardziej lotnego (x) w cieczy (oś X) i (y) w parze (oś Y). Obok linii równowagi prowadzi się pomocniczo przekątną odpowiadająca sytuacji gdy składy cieczy i pary nie różnią się miedzy sobą. Krzywe równowagi dla mieszanin zeotropowej (rys.6) i azeotropowych (rys.7 i rys.8) wyglądają wtedy następująco:
Jak z powyższych rysunków widać, zeotropy to mieszaniny dla których w całym zakresie stężeń skład pary pozostającej w równowadze z cieczą różni się od składu cieczy. zwiększoną zawartością składnika bardziej lotnego. Natomiast azeotropy to mieszaniny, dla których istnieje stężenie, przy którym skład pary nad mieszaniną nie różni się od składu cieczy. W rezultacie mieszaniny azeotropowej prostymi metodami destylacyjnymi nie da się rozdzielić.
Uwzględniając liczbę faz w temperaturze wrzenia mieszaniny ostatecznie rozróżniamy następujące mieszaniny wieloskładnikowe: homozeotropy, homoazeotropy, heterozeotropy i heteroazeotropy.
RODZAJE DESTYLACJI
DESTYLACJA PROSTA
Zasadą prowadzenia tego typu destylacji jest stałe odprowadzanie oparów znad cieczy. W miarę trwania destylacji opary zmieniają swój skład. Wobec odparowywania przede wszystkim składnika lotniejszego w destylowanej cieczy zmniejsza się jego stężenie. W rezultacie, kolejne partie destylatu zawierają coraz mniej składnika lotniejszego. Po zakończeniu procesu skład cieczy wyczerpanej nie jest więc w równowadze ze składem całości otrzymanego destylatu lecz z jego ostatnią frakcją. Przykładem tego typu destylacji jest destylacja w kolbie z ciągłym odbieraniem oparów.
REKTYFIKACJA
Rektyfikacja
polega na przeciwprądowym zetknięciu się cieczy i par z jednoczesną wymianą
masy i ciepła.
W
trakcie procesu podczas kontaktu unoszących się par ze spływającą cieczą
z par wykropleniu ulega mieszanina zubożona o składnik lotny z cieczy zaś
odparowuje mieszanina wzbogacona o składnik lotny. Jednocześnie wydzielone
ciepło kondensacji frakcji wykraplającej się z oparów wykorzystywane jest
do odpędzenia frakcji odparowującej z cieczy. W konsekwencji tych procesów
przechodzące przez kolumnę opary wzbogacają się w składnik najbardziej
lotny, zaś ciecz spływająca w dół kolumny wzbogacana jest wskładnik
mniej lotny.
Pary
dochodzące do szczytu kolumny są skraplane. Warunkiem przeprowadzenia rektyfikacji
jest zawracana części kondensatu z powrotem do kolumny. Oznacza to, że
jedynie część kondensatu odbierana jest jako destylat.
Zawracanie
części kondensatu - t.zw. odcieku - odgrywa w rektyfikacji kluczową rolę.
Stosunek odcieku do destylatu - t.zw. powrót - w istotny sposób wpływa
na parametry procesu. Zwiększenie powrotu z jednej strony zwiększa rozdzielczość
kolumny, z drugiej zwiększa koszty procesu i może powodować trudności techniczne,
np. zalanie kolumny.
Rektyfikację
prowadzi się w kolumnach, które przez swoją konstrukcję spełniają rolę
wielu kotłów umieszczonych jeden nad drugim. Kolumna rektyfikacyjna ma
kształt stojącego walca. Faza ciekła przepływa przez nią grawitacyjnie
z góry na dół, faza gazowa przemieszcza się od dołu do góry wskutek różnicy
ciśnień. Wewnątrz, kolumna wyposażona jest bądź w półki o różnej konstrukcji
bądź zawiera wypełnienie. Wypełnienie stanowią drobne elementy szklane
bądź metalowe - kulki, sprężynki, pocięte rurki.
Konsekwencją
różnic konstrukcyjnych w kolumnach półkowych i z wypełnieniem jest to,
że w kolumnie półkowej zetknięcie faz odbywa się tylko na półkach, podczas
gdy w kolumnie z wypełnieniem kontakt fazy ciekłej i gazowej zachodzi nieprzerwanie
na całej długości kolumny.
Oprócz kolumny zestaw do rektyfikacji obejmuje wyparkę gdzie następuje odparowanie cieczy oraz kondensator, w którym pary z kolumny ulegają skropleniu.
Kryterium podziału procesów rektyfikacyjnych na okresowe i ciągłe jest organizacja procesu. Rektyfikację okresową prowadzi się w sposób następujący: do kotła kolumny (wyparki) wprowadza się mieszaninę ciekłą, która po ogrzaniu do początkowej temperatury wrzenia zaczyna destylować. Z kondensatora na górze kolumny odbiera się kolejne frakcje o coraz wyższych temperaturach wrzenia. W ciągu całego procesu zarówno temperatura skraplania destylatu w kondensatorze jak i temperatura wrzenia mieszaniny znajdującej się w kotle stopniowo rosną. Następuje zatem stopniowe wydzielanie składników mieszaniny począwszy od najbardziej lotnych.
Rektyfikacja okresowa pozwala na rozdzielenie wyjściowej mieszaniny ciekłej zwanej surówką na frakcje różniące się temperaturami kondensacji. Mogą to być czyste związki chemiczne jak również indywidua fizykochemiczne takie jak azeotropy dwu- i wieloskładnikowe.
Rektyfikacja
ciągła polega na nieprzerwanym dozowaniu surówki na kolumnę w czasie trwania
procesu przy równoczesnym odbiorze destylatu z kondensatora i cieczy wyczerpanej
z kotła kolumny. Miejsce na kolumnie, na które doprowadza się surówkę nazywa
się półką zasilaną. Część kolumny powyżej półki zasilanej to część wzmacniająca
(wzbogacająca lub koncentrująca) gdyż pary zawierają tu więcej składnika
lotniejszego niż surowiec. Część kolumny poniżej miejsca zasilania surówką
nazywa się odpędzającą lub wyczerpującą ze względu na stopniowy ubytek
składnika lotniejszego z obu faz.
Wielkością
określającą sprawność kolumny rektyfikacyjnej jest ilość półek teoretycznych
jakie kolumna posiada. Półka teoretyczna jest to pojęcie abstrakcyjne,
oznaczające miejsce w kolumnie, w którym podczas przepływu oparów i odcieku
ustali się równowaga ciecz-para.
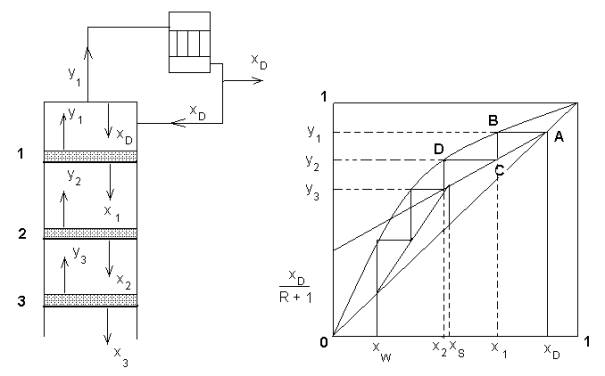
REKTYFIKACJA (UZUPEŁNIENIE)
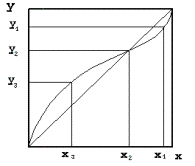
DODATKOWE OBJAŚNIENIA
DO RYS. 2.6 ZAMIESZCZONEGO
- PRACA ZBIOROWA POD REDAKCJĄ T. KASPRZYCKIEJ-GUTTMAN
x1, x2 x3, - ułamki molowe składnika bardziej lotnego w cieczy; y1, y2 y3, - ułamki molowe składnika bardziej lotnego w parze; xS, - ułamek molowy składnika bardziej lotnego w surówce; xD, - ułamek molowy składnika bardziej lotnego w destylacie i odcieku; xW - ułamek molowy składnika bardziej lotnego w cieczy wyczerpanej;
PRZEPAROWANIE
Operacja
jednostkowa, w której do odpędzenia składnika (składników) lotnego używa
się strumienia gazu (najczęściej powietrza) lub pary nazywa się w skrócie
przeparowaniem.
Przeparowanie
zachodzi w kolumnie, w której od góry podawana jest surówka (mieszanina
ciekła co najmniej dwuskładnikowa zawierająca składnik lotny), od dołu
zaś para wodna. Polega ono na przeciwprądowym zetknięciu się cieczy i par
z jednoczesną wymianą masy i ciepła.
W
kolumnie przeparnikowej, podczas kontaktu unoszących się pęcherzyków pary
wodnej ze spływającą z góry kolumny surówką, z cieczy odparowuje mieszanina
wody i składnika lotnego wzbogacona o składnik lotny (LSO) w stosunku do
składu surówki. W rezultacie, w trakcie wędrówki w górę kolumny pęcherzyki
pary wodnej wzbogacają się w składnik lotny, zaś zawartość wody w nich
maleje. Jednocześnie z pęcherzyków pary wykropleniu ulega mieszanina wody
i składnika lotnego zubożona o składnik lotny w porównaniu ze składem pary.
Ciepło
kondensacji frakcji wykraplającej się z oparów wykorzystywane jest do odpędzenia
frakcji odparowującej z cieczy. W konsekwencji tych procesów spływająca
w dół kolumny surówka zubaża się w składnik bardziej lotny, w który wzbogaca
się para wodna.
Pary
dochodzące do szczytu kolumny, wzbogacone w składnik bardziej lotny, są
skraplane. Otrzymany destylat, jeśli składnik lotny słabo rozpuszcza się
w wodzie, rozdziela się na dwie frakcje - wodną i organiczną.
(mgr
Adam Chajewski)
|
|
|