|
WYDZIAŁ
CHEMII - ZAKŁAD DYDAKTYCZNY TECHNOLOGII
CHEMICZNEJ
Ćwiczenie 18 KATALITYCZNE
SPALANIE JAKO METODA OCZYSZCZANIA GAZÓW PRZEMYSŁOWYCH
WSTĘP Niekontrolowany rozwój przemysłu i żywiołowy postęp cywilizacji stanowi poważne zagrożenie dla naturalnego środowiska człowieka. W dniu dzisiejszym najważniejszym problemem jest ochrona środowiska. Jednym z jej aspektów jest utylizacja zanieczyszczeń gazowych emitowanych do atmosfery. Źródłem zanieczyszczeń gazowych obok zakładów przemysłowych są także samochody, kotłownie miejskie i domowe oraz duży udział mają źródła naturalne ( pożary lasów, wybuchy wulkanów, procesy gnilne itp. ). Obecnie problem ograniczenia emisji zanieczyszczeń gazowych jest realizowany w dwóch etapach:
Absorpcja jest to dyfuzyjne przenoszenie cząsteczek substancji z jednej fazy (gazowej) przez granicę faz w objętość drugiej fazy ( cieczy ) wywołane gradientem stężenia w obu fazach. Czyli absorpcjapolega na pochłanianiu zanieczyszczeń gazowych przez ciecz (absorbent). W celu przeniesienia określonej masy zanieczyszczeń z gazu do cieczy konieczne jest przeniknięcie cząstek przez strefę przyległą do granicy faz i przez granicę faz, tj. przez powierzchnię międzyfazową. Przenoszenie cząsteczek do granicy faz zarówno w fazie gazowej, jak i ciekłej określa szybkość dyfuzji cząsteczkowej i burzliwej. Szybkość absorpcji zwiększa się przez zwiększenie powierzchni międzyfazowej oraz zwiększenie szybkości dyfuzji. Zwiększenie powierzchni międzyfazowej można osiągnąć przez rozproszenie jednej fazy w drugiej np. rozproszenie fazy gazowej w cieczy przez zastosowanie bełkotki lub mieszania. Zwiększenie etapu dyfuzji realizuje się przez odpowiednio długi czas zetknięcia faz oraz przez zwiększenie burzliwości przepływu w obu fazach np.: gwałtowne mieszanie. Podczas absorpcji może zachodzić bezprzeponowa wymiana ciepła kondensacja oraz nawilżanie gazów. Jeżeli stężenie zanieczyszczeń jest odpowiednio duże, absorpcja stanowić może metodę odzysku wartościowych substancji. Może stanowić wstępny etap oczyszczania gazu w procesie kompleksowego oczyszczania lub końcowy, gdy absorpcja jest połączona z reakcją chemiczną. Absorpcja stosowana jest wówczas, gdy stężenie zanieczyszczeń wynosi kilka procent a w przypadku gazów rozcieńczonych, gdy są one łatwo rozpuszczalne w absorbencie. Absorbentami są: woda, roztwory kwasów, zasad, soli o właściwościach utleniających lub redukujących. Szybkość absorpcji zwiększa się wówczas, gdy zachodzi reakcja chemiczna między cieczą i gazem, przy czym wzrasta współczynnik wnikania po stronie cieczy Podczas absorpcji z reakcją chemiczną składnik A ze strumienia gazu reaguje z substancją B zawartą w cieczy, w wyniku czego powstaje produkt o właściwościach odmiennych od substancji wyjściowej. Przy oczyszczaniu gazów odlotowych absorpcja z reakcją chemiczną jest jedną z zasadniczych metod usuwania zanieczyszczeń kwaśnych, takich jak SO2, SO3, H2S, NOx, HF, Cl2, HCl i in. Metody absorpcyjne stosowane są często w połączeniu z utlenianiem albo z biodegradacją. W metodach absorpcyjnych połączonych z utlenianiem można stosować jako absorbenty roztwory utleniaczy takich jak chlor, dwutlenek chloru, podchloryn sodowy, nadmanganian potasu oraz obecnie najbardziej popularny ozon. Ozon rozpuszczony jest w absorbencie. Reakcja pomiędzy ozonem i zaabsorbowanym zanieczyszczeniem przebiega bardzo szybko. Jednocześnie następuje likwidacja drobnoustrojów, co często ma duże znaczenie. Zastosowanie ozonu nie powoduje powstawania kłopotliwych odpadów, ponieważ produktem jego rozpadu jest tlen. Niektóre substancje zanieczyszczające gazy odlotowe można skutecznie likwidować na drodze biodegradacji za pomocą mikroorganizmów, które utleniają związki organiczne do dwutlenku węgla i wody lub mineralizują zawarte w nich heteroatomy. Wytwarzana w tym procesie energia jest zużywana przez bakterie. Adsorpcja jest procesem, w którym cząsteczki jednej substancji zostają związane na powierzchni innej substancji, następuje sorpcja zanieczyszczeń gazowych przez stały adsorbent. Adsorpcja polega na wydzielaniu i zatrzymywaniu składników gazu na powierzchni zewnętrznej i wewnętrznej (w porach) ciała stałego. Zatrzymywanie cząsteczek na powierzchni zachodzi w wyniku dziabania sił fizycznych i chemicznych bliskiego zasięgu. Energia wiązania adsorbowanych cząsteczek z powierzchnią jest porównywalna z ciepłem kondensacji. Proces adsorpcji jest egzotermiczny. Proces odwrotny, usuwanie cząsteczek zaadsorbowanych z powierzchni do przestrzeni otaczającego płynu zwany desorpcją wymaga więc doprowadzenia ciepła. Liczba cząsteczek możliwa do zaadsorbowania na powierzchni adsorbentu jest ograniczona i maleje ze wzrostem temperatury. Efektywnej adsorpcji sprzyjać więc będzie duża powierzchnia właściwa adsorbentu i niska temperatura. Adsorpcja jest selektywna i największą zdolność do adsorpcji wykazują cząsteczki gazów o dużej masie i niskiej temperaturze wrzenia. Ulegając adsorpcji, wypierają przy tym inne cząsteczki o mniejszej energii wiązania. Niektóre procesy adsorpcji są nieodwracalne, np. chemisorpcja, dla której energia wiązania cząsteczek na powierzchni jest tu tak duża, że zaadsorbowana substancja może być zdesorbowana tylko w postaci związku chemicznego lub usunięta jak substancja stała. Adsorpcja umożliwia oczyszczanie dużych strumieni gazów o małym stężeniu zanieczyszczeń do poziomu ppm. Podczas adsorpcyjnego oczyszczania gazów zanieczyszczenia o małym stężeniu, 20-30 ppm, są zatężane, co w dalszym etapie umożliwia ich spalenie lub ekonomiczne wykorzystanie. Ponadto tą metodą można jednocześnie usuwać z gazu więcej zanieczyszczeń, zwłaszcza substancje organiczne. Kondensacja jest metodą usuwania z gazów odlotowych substancji o niskim ciśnieniu par w umiarkowanie podwyższonych temperaturach lub o wysokim ciśnieniu par, gdy nie jest wymagane bardzo dokładne oczyszczanie gazu do stężeń kilku ppm. Konieczność wymrażania gazu w końcowym etapie oczyszczania tą metodą ogranicza jej zastosowanie. Spalanie bezpośrednie ( termiczne )
lub katalityczne ) stosowane jest do usuwania z gazów odlotowych węglowodorów
poprzez ich utlenienie do CO2 iH2O.
Jeżeli stężenie węglowodorów w strumieniu gazów odlotowych jest dostatecznie wysokie spala się je najczęściej dozując do palnika zasilanego gazem ziemnym. Jest to tzw. spalanie termiczne. Jest ono bardzo energochłonne i kosztowne. Przeprowadza się je w wysokich temperaturach 800 – 1200oC. Ponadto, w czasie wysokotemperaturowego spalania powstają tlenki azotu na skutek utleniania azotu z powietrza powodujące wtórne zanieczyszczenie atmosfery. Spalanie katalityczne stosuje się w przypadku niskich stężeń węglowodorów w gazach odlotowych. Polega ono na przyspieszeniu reakcji utlenienia przez zastosowanie katalizatorów. W procesie spalania katalitycznego strumień gazu przepuszcza się przez ziarno katalizatora w podwyższonej temperaturze. Katalityczne spalanie węglowodorów przebiega w temperaturach znacznie niższych niż spalanie termiczne, około 250oC. Jako katalizatory spalania węglowodorów stosowane są metale jak: platyna, pallad, ruten, rod lub tlenki metali przejściowych jak: tlenek manganu, chromu, miedzi. Katalizatory stosuje się naniesione na nośniki ceramiczne tzw. adsorbenty. Nośniki charakteryzują się bardzo rozwiniętą powierzchnią wewnętrzną ( kanaliki), mają często kształt plastra miodu. Redukcja katalityczna stosowana w procesach
usuwania tlenków azotu z gazów odlotowych polega na redukcji tlenków azotu
za pomocą amoniaku, tlenku węgla lub węglowodorów w obecności katalizatorów.
Katalizatorami tej reakcji są metale szlachetne jak platyna, pallad, rod naniesione na ceramiczne nośniki. Platyna na nośniku ceramicznym jest katalizatorem stosowanym w samochodach dla dopalania spalin i jednoczesnej redukcji tlenków azotu. Rozkład katalityczny tlenków azotu jest prostą
metodą usuwania ich ze strumienia gazów przemysłowych.
Na dzień dzisiejszy jest to metoda szeroko badana
w laboratoriach i wiąże się z nią duże nadzieje aplikacyjne ponieważ nie
wymaga dodawania do strumienia gazu żadnych reagentów i produktami są tylko
azot i tlen. Najlepszymi katalizatorami tej reakcji są zeolity modyfikowane
jonami miedzi. Zeolity to są krystaliczne glinokrzemiany naturalne i sztuczne
o wzorze ogólnym: M2/nO.
Al2O3.
xSiO2.
yH2O
gdzie x>2,
Usuwanie węglowodorów z gazów na drodze katalitycznego utleniania Narażenie na szkodliwe związki obecne w środowisku jest przyczyną wystepowania wielu chorób w tym nowotworów. Dużo uwagi poświęca się związkom karcenogennym, wsród nich wielopierścieniowym węglowodorom aromatycznym (WWA) ze względu na ich rozpowszechnione występowanie związane z rozwojem cywilizacji. Nie wszystkie WWA są rakotwórcze bądź mutagenne. Międzynarodowa Agencja do Badań nad Rakiem (IARC) w 1983 uznała za rakotwórcze w stosunku do ludzi i zwierząt 30 WWA, między innymi benzo[a]piren i benzo[a]antracen. Amerykańska Agencja Ochrony Środowiska (US EPA) w 1997 uznała 16 WWA za wyjątkowo toksyczne, w związku z czym zaleca się ich oznaczanie w analizie stopnia zanieczyszczenia środowiska. Są to: naftalen, acenaftylen, acenaften, fluoren, fenantren, antracen, fluoranten, piren, benzo[a]antracen, chryzen, benzo[b]fluoranten, benzo[k]fluoranten, benzo[a]piren, dibenzo[a,h]antracen, benzo[g,h,i]perylen, indeno[1,2,3-c,d]piren. Wsród tych 16 węglowodorów znajdują się zarówno WWA powodujace karcenogenezę (benzo[a]piren, dibenzo[a,h]antracen, benzo[b]fluoranten, indeno[1,2,3-c,d]piren) i związki uważane za nie powodujące karcenogenezy (fenantren, antracen, piren, benzo[g,h,i]perylen). Emisja par związków organicznych do atmosfery może pochodzić ze źródeł naturalnych, przemysłowych i wtórnych. Do naturalnych źródeł emisji zalicza się procesy gnilne, pożary lasów i erupcje wulkanów. Do przemysłowych źródeł emisji związków organicznych należą różnorakie procesy technologiczne, z których wydzielają się pary różnych związków organicznych, a w szczególności rozpuszczalników. Wtórne zanieczyszczenie atmosfery związkami organicznymi powodują między innymi spaliny samochodowe oraz pary uchodzące w trakcie magazynowana, transportowania i dystrybucji paliw. W ostatnich latach coraz poważniejszym problemem stają się gazy odlotowe z procesów spalania różnych odpadów komunalnych czy przemysłowych. Obok związków chloroorganicznych występują w nich także wielopierścieniowe węglowodory aromatyczne WWA. Za emisję WWA odpowiedzialne są głównie elektrociepłownie i gospodarstwa domowe (ogrzewanie i gotowanie) (51%), spalanie na wolnym powietrzu (28%) przemysł (np. huty aluminium) (20%) i transport samochodowy (0.9%). Celem oczyszczania gazów odlotowych jest najczęściej nie tyko usunięcie zanieczyszczeń, ale także odzysk rozpuszczalników. Gdy odzysk związków organicznych z gazów jest nieopłacalny należy je usunąc przez bezpośrednie lub katalityczne spalanie. Podstawowe wiadomości o katalizie. Kataliza jest to zjawisko polegające
na zwiększeniu szybkości reakcji chemicznej i/lub skierowaniu reakcji na
jedną z kilku możliwych termodynamicznie dróg prowadzących do różnych produktów,
w obecności niewielkich ilości substancji zwanych katalizatorami.
Substancje te tworząc nietrwałe połaczenia przejściowe, nie sa jednakże
zużywane w reakcji i nie występoują w jej równaniu stechiometrycznym. Katalizator
nie zmienia przy tym położenia równowagi chemicznej, wpływa jedynie na
szybkość dochodzenia układu do tego stanu.
Im szybciej katalizator doprowadza daną reakcję do
stanu równowagi tym jest aktywniejszy. Aktywność katalizatora Ak
określa się jako różnicę między szybkościami reakcji chemicznej zachodzącej
w obecności katalizatora, vk,
i bez niego, vh:
Szybkość reakcji bez katalizatora jest znikomo mała w porównaniu z reakcją katalizowaną stąd miarą aktywności katalizatora jest wprost szybkość reakcji vk. Aktywność katalizatora ma sens tylko w odniesieniu do układu katalizator-reagenty, ma ona sens tylko w odniesieniu do danego typu reakcji np: aktywność katalizatora tlenku wanadu w reakcji utlenienia węglowodorów aromatycznych lub w reakcji utlenienia SO2 do SO3. Katalizator im bardziej przyspiesza określoną reakcję danego substratu tym jest bardziej selektywny. Selektywny katalizator przyspieszając przebieg reakcji tylko w jednym kierunku pozwala otrzymać produkt o dużej czystości, przy małym jednostkowym zużyciu surowców. Selektywność katalizatora Sidefiniuje się jako stosunek ilości cpi jednego z kilku możliwych produktów (Pi) reakcji do całkowitej ilości produktów S cpi:
W praktyce przemysłowej selektywność = 70-90%, dla enzymów = 100%, Ze względów praktycznych reakcje katalityczne dzieli się na dwie odrębne kategorie. Gdy katalizator znajduje się w tej samej fazie co substraty i nie występuje granica faz, mówimy o katalizie homogenicznej, czyli jednofazowej. Gdy katalizator stanowi odrębną fazę mówimy o katalizie heterogenicznej. Najczęściej stosowanym układem heterofazowym jest układ w którym katalizator jest stały natomiast substrat występuje w fazie gazowej. Trzecią grupą katalizatorów, nie mieszczącą się w podanej klasyfikacji są enzymy. Są to duże złożone cząstki organiczne, które nie są układem homogenicznym ani heterogenicznym lecz czymś pośrednim. Kataliza heterogeniczna. Zdecydowana większość najważniejszych procesów przemysłu chemicznego prowadzona jest w obecności katalizatorów stałych tzw. kontaktów, głównie ze względu na łatwość oddzielenia katalizatora od mieszaniny reakcyjnej. Proces katalizy na stałych katalizatorach porowatych składa się z następujących etapów:
Stałą szybkości procesu heterogenicznego ( k*) opisuje następujące wyrażenie (.1 ):
D - współczynnik dyfuzji przez warstewkę laminarną o grubości z. Gdy odwrotności odpowiednich stałych szybkości uzna
się za miarę oporów poszczególnych etapów, to równanie (2) wskazuje, że
opór procesu heterogenicznego 1/k* równy jest sumie oporów: kinetycznego
1/k i dyfuzyjnego 1/(d/z). Całkowity opór procesu heterogenicznego opisuje
następujące wyrażenie:
Jeśli wartość stałej szybkości reakcji chemicznej k znacznie przewyższa wartość współczynnika wnikania masy D/z ( k >>D/z) to opór dyfuzji określa szybkość przemiany i proces przebiega w obszarze dyfuzyjnym. Natomiast gdy współczynnik wnikania masy D/z jest bardzo duży w porównaniu ze stałą szybkości reakcji chemicznej (D/z>> k) szybkość przemiany uwarunkowana jest oporem reakcji chemicznej. Jest to tzw. obszar kinetyczny przebiegu reakcji. Ze wzrostem temperatury w układzie obserwuje się przejście od obszaru kinetycznego do obszaru dyfuzyjnego procesu (pod warunkiem, że pozostałe parametry wpływające na szybkość przemiany nie ulegają zmianie). Jeżeli stadium najwolniejszym ograniczającym szybkość procesu jest dyfuzja to najskuteczniejszym sposobem jego przyspieszenia jest zwiększenie ruchu burzliwego strumienia gazu (cieczy) w złożu nieruchomym katalizatora lub mieszanie mieszaniny reakcyjnej z katalizatorem. Proces zachodzi w obszarze dyfuzji wewnętrznej, gdy najwolniejsze są stadia b i f. Najskuteczniejszym sposobem intensyfikacji w tym przypadku jest zmniejszenie średnicy ziaren katalizatora i zwiększenie wymiaru porów. Wzrost stężenia reagentów lub ciśnienia przyspiesza zarówno dyfuzyjne jak i kinetyczne stadia katalizy. Dla procesów przebiegających w obszarze kinetycznym zwiększenie temperatury zawsze przybliża proces do stanu równowagi, lecz wydajność równowagowa ze wzrostem temperatury zwiększa się w przypadku procesów endotermicznych i maleje dla procesów egzotermicznych. Zastosowanie ciśnienia jest jednym ze sposobów zwiększenia wydajności odwracalnych reakcji katalitycznych na skalę przemysłową, przebiegających ze zmniejszeniem objętości produktów gazowych. Katalizatory heterogeniczne stanowią jedno- lub wielofazowe układy ciał stałych o zróżnicowanym składzie chemicznym. Formowane są w postaci nieregularnych lub o określonych kształtach ziaren i ze względu na możliwość zapewnienia jak największego kontaktu z reagentami preparuje się je by miały one jak największą powierzchnię właściwą. Otrzymuje się to poprzez wytworzenie porowatej struktury ziarna. Jako, że powierzchnia ścian porów jest nawet kilkaset razy większa od powierzchni zewnętrznej ziaren, reakcja biegnie przede wszystkim wewnątrz ziaren katalizatora. Do katalizatora w trakcie wytwarzania dodaje się różne substancje zwane promotorami. Promotory mogą zmieniać własności kontaktu, nazywa się je wtedy promotorami chemicznymi. Promotory mogą zmieniać strukturę wewnętrzną kontaktu oraz jego powierzchnię - są to promotory strukturalne. Często aktywny składnik katalizatora osadza się na składniku nieaktywnym aby zwiększyć powierzchnię kontaktu lub ułatwić odprowadzenie ciepła wytworzonego w czasie reakcji. Taki nieaktywny składnik nazywa się nośnikiem. Podczas katalizy heterogenicznej reakcja przyśpieszana jest poprzez tworzenie na powierzchni katalizatora przejściowego kompleksu aktywnego z reagentami. Jedynie pewne fragmenty powierzchni wykazują aktywność katalityczną. Są to wzniesienia. naroża, krawędzie mikrokryształów lub inne defekty mikrostrukturalne powierzchni. Na tych fragmentach powierzchni, zwanych centrami aktywnymi następuje adsorpcja a potem chemisorpcja substratów. Aktywność katalizatora ściśle zależy od chemisorpcji substratu na powierzchni. Jeśli substrat jest bardzo silnie chemisorbowany nie opuści centrum aktywnego katalizatora. Efekt ten nazywamy zatruciem. W przypadku bardzo słabej adsorpcji oddziaływanie katalizatora na reagenty jest zbyt słabe i substraty szybciej desorbują niż wytwarzają produkty. Jednym z ważniejszych problemów katalizy heterogenicznej jest zatrucie katalizatora, które polega na częściowej lub całkowitej utracie aktywności wskutek działania niewielkich ilości substancji zwanych truciznami kontaktu. Trucizny zwykle są wprowadzane wraz z surowcami wejściowymi. Zatrucie następuje wskutek trwałej adsorpcji lub reakcji trucizny z katalizatorem i powstania związku katalitycznie nieaktywnego. Zatrucie może być odwracalne i nieodwracalne. Szczególnie wrażliwe są na zatrucia katalizatory metaliczne. Truciznami katalizatorów są: siarkowodór, siarczki organiczne i nieorganiczne, związki arsenu, fosforowodór, amoniak. Oprócz zatrucia aktywność katalizatorów może maleć z powodu:
Vm = objętość mieszaniny reakcyjnej przepływającej przez katalizator (m3 /s) Wpływ czasu zetknięcia t jest podobny dla wielu reakcji katalitycznych. Z przedłużaniem czasu kontaktu wydajność produktu odwracalnej reakcji katalitycznej wzrasta, natomiast intensywność pracy aparatu kontaktowego G/t spada. ( G = ilość produktu w jednostce czasu z jednostki objętości katalizatora). Im aktywniejszy katalizator ( A1> A2> A3 ) tym krótszy jest czas zetknięcia ( t1< t2< t3 ) potrzebny do uzyskania założonej wydajności produktu x. Katalizatory heterogenicznych reakcji utleniania-redukcji Katalizatorami tej grupy reakcji są przewodniki prądu elektrycznego - metale oraz półprzewodniki - tlenki i siarczki metali. Największą aktywnością i róźnorodnością działania odznaczają się metale długich okresów układu okresowego pierwiastków. Są to w zasadzie metale I,VI,VII,VIII grupy: Cu, Ag, Cr, Mo, W, U, Fe, Co, Ni, Pt, Pd itp. Wszystkie wymienione metale są pierwiastkami przejściowymi o niezapełnionej powłoce d i charakteryzujące się takimi własnościami jak:zmienna wartościowość, podatność do tworzenia kompleksów, stosunkowo niewielką energią potrzebną do wyrwania elektronu. Wysunięto przypuszczenie, że wolne i zapełnione orbitale d są potrzebne do luźnego związania adsorbowanej cząsteczki z powierzchnią w fazie wstępnej, z której cząsteczka ta przechodzi później w stan trwałego związania z powierzchnią. Katalizatory utleniania węglowodorów Katalizatory zawierające platynę i pallad są bardziej aktywne w procesie pełnego utlenienia węglowodorów, szczególnie aromatycznych od katalizatorów, w których skład wchodzą tlenki metali Cu, Mn, Cr. Fe, Co, Sn, Zn. Ze względu na wysoką cenę metali aktywnych katalitycznie Pt, Pd, w przemyśle stosuje się metale osadzone na nośniki nieorganiczne. Jako nośniki katalityczne mogą być stosowane drobnoziarniste materiały ceramiczne lub nośniki monolituczne o strukturze komórkowej, na bazie tlenku glinu lub glinokrzemianów. Dobry nośnik powinien charakteryzować się następującymi własnościami:
Obecnie wykorzystywane nośniki można podzielic na dwie grupy:
Instrukcja wykonania ćwiczenia.
Celem ćwiczenia jest przedstawienie metody katalitycznego utleniania węglowodorów jako wysoce wydajnej metody oczyszczania gazów. W ćwiczeniu zastosoawny jest katalizator platyna naniesiona na tlenek glinu. Jako przykład substancji stanowiącej zanieczyszczenie powietrza wybrano heksan. Opis ćwiczenia.
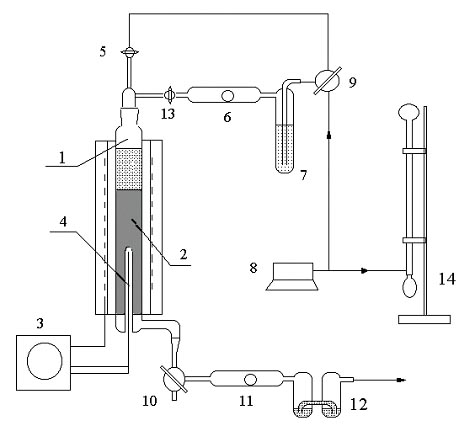
Schemat aparatury
Wymagania
Proponowana literatura uzupełniająca:
|
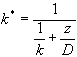 (1)
(1) 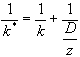 (2)
(2)