|
WYDZIAŁ
CHEMII - ZAKŁAD DYDAKTYCZNY TECHNOLOGII
CHEMICZNEJ
ĆWICZENIE 26 Kinetyka reakcji dehydratacji wobec katalizatora heterogenicznego (Al2O3) Kataliza – wiadomości wstępne Kataliza jest to zjawisko polegające na zwiększeniu szybkości reakcji chemicznej i/lub skierowaniu reakcji na jedną z kilku możliwych termodynamicznie dróg prowadzących do różnych produktów, w obecności niewielkich ilości substancji zwanych katalizatorami. Substancje te tworząc nietrwałe połaczenia przejściowe, nie sa jednakże zużywane w reakcji i nie występoują w jej równaniu stechiometrycznym. Katalizator nie zmienia przy tym położenia równowagi chemicznej, wpływa jedynie na szybkość dochodzenia układu do tego stanu. Katalizator definiuje się więc jako substancję, która zwiększa szybkość z jaką reakcja chemiczna osiąga stan równowagi, sama się jednak nie zużywa i której symbol nie występuje w równaniu stechiometrycznym. Wyjątkiem jest zjawisko przyspieszania reakcji przez produkty zwane autokatalizą. Wpływ katalizatora na reakcję chemiczną polega głównie na obniżeniu jej energii aktywacji. Potwierdza to wiele faktów doświadczalnych. Tłumaczy się to tworzeniem przejściowych aktywnych kompleksów katalizatora z substratami, po rozpadzie których katalizator ulega regeneracji. Należy zaznaczyć, że położenie równowagi osiągnięte w obecności katalizatora jest identyczne z połoźeniem, do którego ostatecznie dochodzi układ bez katalizatora oraz, że katalizator może jedynie zwiększyć szybkość reakcji, która jest termodynamicznie moźliwa, nie może natomiast zapoczątkować reakcji, która jest termodynamicznie niemożliwa. Im szybciej katalizator doprowadza daną reakcję do stanu równowagi tym jest aktywniejszy.Aktywność katalizatora Ak określa się jako różnicę między szybkościami reakcji chemicznej zachodzącej w obecności katalizatora, vk, i bez niego, vh:
Szybkość reakcji bez katalizatora jest znikomo mała w porównaniu z reakcją katalizowaną stąd miarą aktywności katalizatora jest wprost szybkość reakcji v k. Aktywność katalizatora ma sens tylko w odniesieniu do układu katalizator-reagenty, ma ona sens tylko w odniesieniu do danego typu reakcji np: aktywność katalizatora tlenku wanadu w reakcji utlenienia węglowodorów aromatycznych lub w reakcji utlenienia SO2 do SO3.
Szybkość reakcji katalitycznej można odnieść do jednostki masy, objętości, powierzchni katalizatora lub liczby miejsc aktywnych. W zależności od odniesienia wyraża się jako:
TON – turnover number ( ilość cykli katalitycznych ) – liczba moli substratu ulegającego reakcji w przeliczeniu na mol katalizatora [ mol.mol-1]; do powierzchni właściwej katalizatora [mol.m -2.s-1]do jednostki masy katalizatora [mol.g-1.s-1] do objętości katalizatora {mol.cm -3. s-1]. Katalizator im bardziej przyspiesza określoną reakcję danego substratu tym jest bardziej selektywny. Selektywny katalizator przyspieszając przebieg reakcji tylko w jednym kierunku pozwala otrzymać produkt o dużej czystości, przy małym jednostkowym zużyciu surowców. Selektywność katalizatora S definiuje się jako stosunek ilości n pi jednego z kilku możliwych produktów (Pi) reakcji do całkowitej ilości produktów S npi:S X ® P 1 + P2 + P3 + ...+Pi
W praktyce przemysłowej selektywność = 70-90%, enzymy 100%, Ze względów praktycznych reakcje katalityczne dzieli się na dwie odrębne kategorie. Gdy katalizator znajduje się w tej samej fazie co substraty i nie występuje granica faz, mówimy o katalizie homogenicznej, czyli jednofazowej. Gdy katalizator stanowi odrębną fazę mówimy o katalizie heterogenicznej. Najczęściej stosowanym układem heterofazowym jest układ w którym katalizator jest stały natomiast substrat występuje w fazie gazowej.Trzecią grupą katalizatorów, nie mieszczącą się w podanej klasyfikacji są enzymy. Są to duże złożone cząstki organiczne, które nie są układem homogenicznym ani heterogenicznym, lecz czymś pośrednim.
Wszystkie procesy katalityczne można podzielić pod względem mechanizmu reakcji na:
Kataliza heterogeniczna. Zdecydowana większość najważniejszych procesów przemysłu chemicznego prowadzona jest w obecności katalizatorów stałych tzw. kontaktów, głównie ze względu na łatwość oddzielenia katalizatora od mieszaniny r eakcyjnej.Proces katalizy na stałych katalizatorach porowatych składa się z następujących etapów: dyfuzja reagentów w porach ziaren katalizatora do jego powier adsorpcja reagentów na powierzchni katalizatora; reakcja chemiczna na powierzchni katalizatora; desorpcja produktów reakcji z powierzchni katalizatora; dyfuzja produktów w porach ziaren katalizatora; dyfuzja produktów reakcji od powierzchni katalizatora do strumienia gazu.
Szybkość ogólna heterogenicznego procesu katalitycznego określana jest względnymi szybkościami poszczególnych stadiów i jest ograniczona poprzez stadium najwolniejsze. Etap d) reakcja chemiczna jest przyspieszana przede wszystkim przez podwyższenie temperatury. Wraz ze wzrostem temperatury stała szybkości reakcji chemicznej rośnie znacznie szybciej niż współczynnik dyfuzji. Stałą szybkości procesu heterogenicznego ( k*) opisuje następujące wyrażenie (1 ):
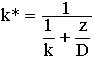 (1) (1)
k - stała szybkości etapu reakcji chemicznej, D - współczynnik dyfuzji przez warstewkę laminarną o grubości z. Gdy odwrotności odpowiednich stałych szybkości uzna się za miarę oporów poszczególnych etapów, to równanie (1) wskazuje, że opór procesu heterogenicznego równy jest sumie oporów: kinetycznego 1/k i dyfuzyjnego 1/(D/z).
Jeśli wartość stałej szybkości reakcji chemicznej k znacznie przewyższa wartość współczynnika wnikania masy D/z ( k >>D/z) to opór dyfuzji określa szybkość przemiany i proces przebiega w obszarze dyfuzyjnym. Natomiast gdy współczynnik wnikania masy D/z jest bardzo duży w porównaniu ze stałą szybkości reakcji chemicznej (D/z>> k) szybkość przemiany uwarunkowana jest opo rem reakcji chemicznej. Jest to tzw. obszar kinetyczny przebiegu reakcji.Ze wzrostem temperatury w układzie obserwuje się przejście od obszaru kinetycznego do obszaru dyfuzyjnego procesu (pod warunkiem, że pozostałe parametry wpływające na szybkość przemiany nie ulegają zmianie). Jeżeli stadium najwolniejszym ograniczającym szybkość procesu jest dyfuzja to najskuteczniejszym sposobem jego przyspieszenia jest zwiększenie ruchu burzliwego strumienia gazu (cieczy) w nieruchomym złożu katalizatora lub m ieszanie mieszaniny reakcyjnej z katalizatorem. Proces zachodzi w obszarze dyfuzji wewnętrznej, gdy najwolniejsze są stadia b i f. Najskuteczniejszym sposobem intensyfikacji w tym przypadku jest zmniejszenie średnicy ziaren katalizatora i zwiększenie rozmiaru porów. Wzrost stężenia reagentów lub ciśnienia przyspiesza zarówno dyfuzyjne jak i kinetyczne stadia katalizy. Dla procesów przebiegających w obszarze kinetycznym zwiększenie temperatury zawsze przybliża proces do stanu równowagi, lecz wydajność równowagowa ze wzrostem temperatury zwiększa się w przypadku procesów endotermicznych i maleje dla procesów egzotermicznych. Zastosowanie ciśnienia jest jednym ze sposobów zwiększenia wydajności odwracalnych reakcji katalitycznych na skalę przemysłową, przebiegających ze zmniejszeniem objętości produktów gazowych.Katalizatory heterogeniczne stanowią jedno- lub wielofazowe układy ciał stałych o zróżnicowanym składzie chemicznym. Formowane są w postaci ziaren o nieregularnych lub określonych kształtach i ze względu na możliwość zapewnienia jak największego kontaktu z reagentami preparuje się je tak by miały jak największą powierzchnię właściwą. Otrzymuje się to poprzez wytworzenie porowatej struktury ziarna. Jako, że powierzchnia ścian porów jest nawet kilka set razy większa od powierzchni zewnętrznej ziaren, reakcja biegnie przede wszystkim wewnątrz ziaren katalizatora.Do katalizatora w trakcie wytwarzania dodaje się różne substancje zwane promotorami. Promotory mogą zmieniać własności kontaktu - nazywa się je wtedy promotorami chemicznymi. Takim promotorem jest tlenek potasu w żelazowym katalizatorze do syntezy amoniaku. Promotory mogą zmieniać strukturę wewnętrzną kontaktu oraz jego powierzchnię - są to promotory struk turalne. Często aktywny składnik katalizatora osadza się na składniku nieaktywnym aby zwiększyć powierzchnię kontaktu lub ułatwić odprowadzenie ciepła wytworzonego w czasie reakcji. Taki nieaktywny składnik nazywa się nośnikiem.Podczas katalizy heteroge nicznej reakcja przyśpieszana jest poprzez tworzenie na powierzchni katalizatora przejściowego kompleksu aktywnego z reagentami. Jedynie pewne fragmenty powierzchni wykazują aktywność katalityczną. Są to wzniesienia, naroża, krawędzie mikrokryształów lub inne defekty mikrostrukturalne powierzchni. Na tych fragmentach powierzchni, zwanych centrami aktywnymi następuje adsorpcja, a potem chemisorpcja substratów. Centra aktywne można rozpatrywać jako grupy atomów najsłabiej związane z powierzchnią i znajdujące się w najkorzystniejszych warunkach do tworzenia wiązań i wymiany elektronów lub protonów wskutek występowania nienasyconych wiązań walencyjnych w tych atomach.Aktywność katalizatora ściśle zależy od chemisorpcji substratu na powierzchni. Jeśli substrat jest bardzo silnie chemisorbowany to nie opuści centrum aktywnego katalizatora. Efekt ten nazywamy zatruciem. W przypadku bardzo słabej adsorpcji oddziaływanie katalizatora na reagenty jest zbyt słabe i substraty szybciej desorbują niż wytwarzają produkty. Jednym z ważniejszych problemów katalizy heterogenicznej jest zatrucie katalizatora, które polega na częściowej lub całkowitej utracie aktywności wskutek działania niewielkich ilości substancji zwanych truciznami kontaktu. Trucizny zwykle są wprowadzane wraz z surowcami wejściowymi. Zatrucie następuje wskutek trwałej adsorpcji lub reakcji trucizny z katalizatorem i powstania związku katalitycznie nieaktywnego. Zatrucie może być odwracalne i nieodwracalne. Szczególnie wrażliwe są na zatrucia katalizatory metaliczne. Truciznami katalizatorów są: siarkowodór, siarczki organiczne i nieorganiczne, związki arsenu, fosforowodór, amoniak.Oprócz zatrucia aktywność katalizatorów może maleć z powodu: zmniejszenia powierzchni aktywnej przy mechanicznym pokrywaniu powierzchni katalizatora zanieczyszczeniami np: pyłem lub substancjami stałymi powstającymi podczas katalizy. Takim przykładem blokowania powierzchni jest gromadzenie się związków węgla na powierzchni glinokrzemianów podczas krakingu katalitycznego. Zregenerowanie katalizatora następuje przez wypalenie powstałego koksu.
Czas kontaktu reagentów z katalizatorem jest ważną charakterystyką procesu technologicznego, która umożliwia obliczanie objętości reaktorów. Czas kontaktu można obliczyć ze wzoru: Wpływ czasu zetknięcia t jest podobny dla wielu reakcji katalitycznych. Z przedłużaniem czasu kontaktu wydajność produktu odwracalnej reakcji katalitycznej wzrasta, natomiast intensywność pracy aparatu kontaktowego G/t spada. ( G = ilość produktu w jednostce czasu z jednostki objętości katalizatora). Im aktywniejszy katalizator ( A 1> A2> A3 ) tym krótszy jest czas zetknięcia (t 1< t 2< t 3 ) potrzebny do uzyskania założonej wydajności produktu x.
Katalizatory heterogenicznych reakcji utleniania-redukcji. Katalizatorami procesów utleniania-redukcji n p: uwodornienia, utlenienia są przewodniki prądu elektrycznego - metale oraz półprzewodniki - tlenki i siarczki metali. Największą aktywnością i różnorodnością działania odznaczają się metale długich okresów układu okresowego pierwiastków Są to w zasadzie metale I,VI,VII,VIII grupy: Cu,Ag,Cr,Mo,W,U,Fe,Co,Ni,Pt,Pd itp. Wszystkie wymienione metale są pierwiastkami przejściowymi o niezapełnionej powłoce d i charakteryzujące się takimi własnościami jak: zmienna wartościowość, podatność do tworzenia kompleksów, stosunkowo niewielką energią potrzebną do wyrwania elektronu.
Katalizatory heterogenicznych reakcji kwasowo-zasadowych. Do grupy reakcji, zwanych katalizą kwasowo-zasadową lub jonową należą reakcje izomeryzacji, krakingu, alkilowania, polimeryzacji przebiegające wobec glinokrzemianów (Al 2O3-SiO2) jako katalizatorów oraz odwodnienie na katalizatorach, którymi są tlenki izolatory takie jak Al2O3, SiO2, MgO. Cechą charakterystyczną katalizatorów glinokrzemianowych ( naturalnych i syntetycznych) są ich własności kwasowe. Wynikają one z zakłóceń sieci krystalicznej krzemionki spowodowanych wprowadzeniem atomów glinu w miejsce atomów krzemu. Podstawiając krzem glinem otrzymuje się jonregularna struktura krystaliczna i stała średnica kanalików co umożliwia reagowanie jedynie cząsteczkom o rozmiarach nie przekraczających pewnej granicy, obecność silnie kwasowych grup wodorotlenowych, które mogą zainicjować reakcje z udziałem karbokationów, obecność bardzo silnych pól elektrostatycznych w sąsiedztwie kationów, które mogą wzbudzić reaktywność w cząsteczkach substratu.
Kataliza homogeniczna. Klasyczną katalizą homogeniczną jest kataliza kwasowo-zasadowa w roztworach, zaś klasycznym katalizatorem tej reakcji jest proton ( Reakcje katalityczne w homofazie mają szereg zalet. Można je prowadzić w niskich temperaturach i przy niskich ciśnieniach, tak aktywne są kompleksowe katalizatory. Charakteryzują się też bardzo dużą selektywnością. Zasadniczą wadą katalizy homogenicznej jest trudność wyodrębnienia katalizatora z mieszaniny poreakcyjnej wskutek czego część katalizatora traci się bezużytecznie a produkt pozostaje nim zanieczyszczony. Pomimo tych niedogodności kompleksy metali przejściowych są coraz szerzej stosowane w przemyśle ze względu na dużą wydajność reakcji.
Kataliza enzymatyczna. Enzymy są stereospecyficznymi, biologicznymi katalizatorami podlegającymi tym samym kinetycznym i termodynamicznym ograniczeniom jak katalizatory chemiczne. Enzymy są katalizatorami o wielkiej mocy działania z czterech powodów:
Zakres reakcji katalizowanych przez enzymy jest o wiele szerszy niż reakcji katalizowanych nieenzymatyczne. Enzymy są zwykle bardzo specyficzne - dany enzym katalizuje tylko jeden typ reakcji. Enzymy katalizują reakcje substratów o określonej strukturze i powodują powstawanie produktu o określonej strukturze. Dzięki temu moźe on wybiórczo przekształcać jeden składnik mieszaniny w jednen produkt o 100% czystości. Zmiany własności enzymów są bardzo zależne od sposobu syntezy, degradacji jak i moźliwości reakcji z małymi cząsteczkami, które aktywują jego działanie.
Kataliza enzymatyczna jeszcze dzisiaj jest bardzo kosztowna ale ze względu na przedstawione korzyści znajduje ona zastosowanie w przemyśle spoźywczym i farmaceutycznym i stanowi 10% udziału stosowania katalizatorów na świecie. Opanowanie syntez i stosowania katalizatorów enzymatycznych będe miało wielkie znaczenie dla problemów ekologicznych, źywnościowych i energetycznych św iata np: enzymatyczna fotoliza wody, enzymatyczna produkcja skrobi, białka itp.
Dehydratacja alkoholi na katalizatorze stałym ( Al 2O3)Dehydratacja etanolu do etylenu jest historycznie pierwszym przykładem przeprowadzenia reakcji katalitycznej na kontakcie stałym. W roku 1787 dokonał tego Priestley, przepuszczając pary etanolu przez rozgrzaną rurkę kaolinową ale nie wyjaśnił tego zjawiska. Dopiero póżniejsze badania dowiodły, że czynnikiem odwadniającym był tlenek glinu zawarty w kaolinie. Stwierdzono, że reakcja przebiega w temp. 300-400oC i obok olefin powstają etery i aldehydy jako produkty uboczne.Odwodnienie alkoholi do olefin jest metodą prostę, ale kosztowną i stosowaną w skali przemysłowej tylko w krajach, w których inne źródła aklenów są niedostępne czy też niewystarczające. Proces dehydratacji przeprowadza się w fazie gazowej w temperaturze 350-400 oC, pod ciśnieniem atmosferycznym wobec katalizatorów takich jak tlenki glinu, toru, chromu oraz boksyt).Al 2O3 dla celów katalizy prepatowany jest w postaci uwodnionej, którą poddaje się kalcynacji w temperaturze powyżej 200oC. W niższych temperaturach powierzchnia tlenku glinu całkowicie pokryta jest grupami hydroksylowymi. W czasie ogrzewania następuje dehydroksylacja z wydzieleniem wody i tworzeniem jonów tlenkowych O2-.. Bez defektów przestrzennych może zostac usunięte około 68% powierzchniowych grup OH. Dalsza dehydroksylacja prowadzi do powstawania defektów powierzchniowych generujących centra kwasowe Lewisa ( nienasycone koordynacyjnie jony Jon karbonowy na powierzchni k atalizatora rozpada się, dając olefinę, a proton przechodzi do katalizatora Proponowany jest także następujący mechanizm reakcji przez tworzenie wiązania z powierzchniowymi atomami glinu – schemat 1.
Schemat 1. W prowadzenie do ćwiczenia 26.
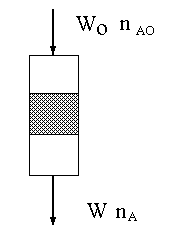
W ćwiczeniu badamy reakcję dehydratacji heksanolu wobec tlenku glinu jako katalizatora. Reakcja jest prowadzona w sposób ciągły w izotermicznym reaktorze ze stałym złożem katalizatora. Alkohol w fazie gazowej przepływa z góry do dołu. Dla tego sposobu prowadzenia reakcji charakterystyczne jest, że gdy warunki reakcji ustabilizują się, stopień przemiany nie zmienia się w czasie. Zależy on od czasu przebywania substratu w reaktorze.
Przez złoże katalizatora przepływa gaz jak pok azano na rysunku.
Przebiega reakcja:
Na wlocie do reaktora prędkość objętościowa heksanolu wynosi W o [ml/h], a ilość moli heksanolu jest no. Zmiana molowości w równaniu (aA = bB + cC ) wynosi m = b + c – a w naszym przypadku 1. Na wylocie ilość moli heksanolu wynosi n. Stopięń konwersjia = (n o – n)/no (5)szybkość reakcji ( sumaryczna szybkość procesu heterogenicznego) wyrażamy prędkością zmian stężenia substratu: r = - dc/dt = k*c (6)
W wyniku całkowania w założonych granicach otrzymamy zależność:
t = 1/k* ln C o/C (7)Reakcja dehydratacji heksanolu jest reakcją pierwszego rzedu przebiegającą ze zmianą molowości tzn. z jednej cząsteczki substratu powstają 2 cząsteczki produktów. Na wylocie reaktora mieszanina poreakcyjna składa się z n moli nieprzereagowanego heksanolu n = n o (1- a ) oraz noa moli heksenu i noa moli wody. Suma moli w mieszaninie wychodzącej wynosi:n o (1- a ) + no a + noa = no (1+ a ) (8)a stężenie hekanolu można przedstawić ułamkiem: n o (1- a )/ no (1+ a ) = (1- a )/ (1+ a ) (9)Podstawiając do wzoru (7) wyrażenia na stężenie w strumieniu wychodzącym otrzymamy: t = 1/k* ln (1 + a )/(1 - a ) (10) Zależność (10) pozwala obliczyć średni czas przebywania reagentów w reaktorze konieczny dla uzyskania stopnia konwersji a . Czas przebywania w reaktorze można obliczyć z równania (11): Vk = objętość katalizatoraVo = szybkość objętościowa substratu w jednostakch objętości na jednostkę czasu.Obliczony czas z równania (11) podstawiamy do równania (10) a znając stpień przemiany a możemy obliczyć pozorną stałą szybkości reakcji dehydratacji heksanolu – k*. Tak wyliczona pozorna stała szybkości jest funkcją stężenia centrów aktywnych na powierzchni katalizatora, adsorpcji substratów na powierzchni, desorpcji produktów z powierzchni katalizatora. Rzeczywista szybkość reakcji chemicznej na powierzchni kontaktu j est wyrażona równaniem:rrz = k c* c sk - rzeczywista stała szybkości reakcji chemicznej na powierzchni katalizatora, c* - stężenie centrów aktywnych na powierzchni, c s – stężenie zaadsorbowanego substratu (heksanolu).Ćwiczenie 26 Kinetyka reakcji dehydratacji wobec katalizatora heterogenicznego (Al2O3)
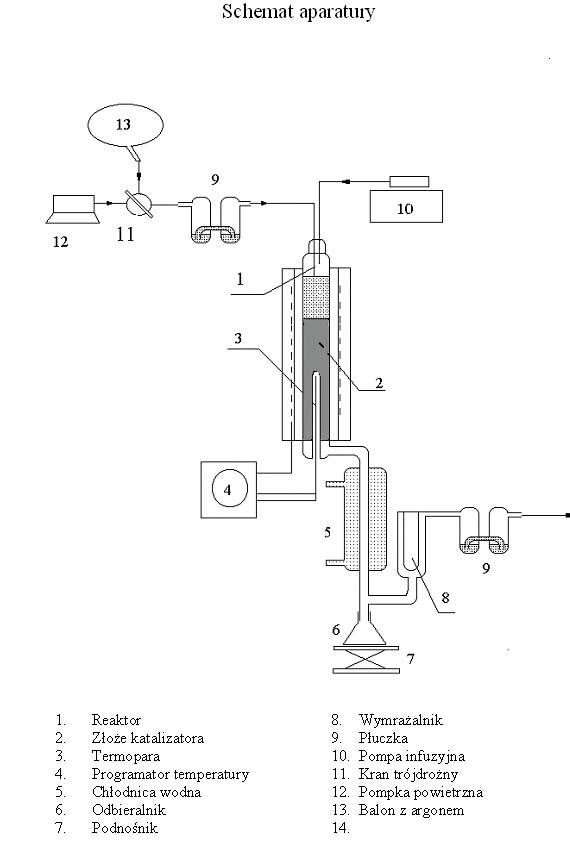
Cel ćwiczenia. Celem ćwiczenia jest zapoznanie z procesem heterogenicznej katalizy oraz z metodami określania parametrów kinetycznych procesu takich jak: czas przebywania substratów w reaktorze, czas kontaktu, stopień przemiany i pozorna stała szybkości reakcji. Jako modelową wybrano do badań reakcję odwodnienia alkoholu wobec tlenku glinu jako katalizatora. Reakcja jest prowadzona w izotermicznym, przepływowym reaktorze z nieruchomą warstwą katalizatora. Jako metoda analizy produktów reakcji jest zastosowana chromatografia gazowa. Analizę prowadzi się na aparacie Hewllet-Packard GC 6890. Zestaw aparatury. Na schemacie przedstawiono zestaw aparatury dla ćwiczenia 26. Reakcja prowadzona jest w izotermicznym, reaktorze (1) z nieruchomym złożem katalizatora (2). Temperatura wewnątrz reaktora jest mierzona za pomocą termopary (3) i kontrolowana za pomocą programatora temperatury (4). Alkohol jest dozowany do reaktora za pomocą strzykawki zamontowanej na pompie infuzyjnej (10). Produkty reakcji przechodząc przez chłodnicę (5) schładzają się i skraplają w odbieralniku (6). Wymrażalnik (8) wypełniony “suchym lodem” zapewnia wykroplenie resztek poreakcyjnych. Płuczki (9) wskazują czy przepływa gaz. Pompka powietrzna (12) pompuje powietrze na złoże katalizatora w czasie regeneracji tlenku glinu. Argon jest stosowany do przepłukania złoża katalizatora po reakcji. Wykonanie ćwiczenia. Zestaw aparatury jak na załączonym rysunku. W reaktorze (1) znajduje się 5ml (4,6g) katalizatora Al2O3 (2). Załączyć grzanie pieca do określonej przez asystenta prowadzącego temperatury i zregenerować katalizator przepuszczając powietrze w temp 600 Zważyć odbieralnik (6). Obniżyć temperaturę pieca do temperatury podanej przez asystenta ( 250-350 Nastawić odpowiednie prędkości przepływu na pompie infuzyjnej (10). Napełnić strzykawkę alkoholem i zamontować na pompie. Podłączyć odbieralnik (6) na dole chłodnicy. Zanotować godzinę i rozpocząć dozowanie alkoholu. Proces jest prowadzony przez czas określony przez asystenta. Po upływie określonego czasu należy zważyć odbieralnik i wykonać analizę produktów na chromatografie gazowym. Obliczyć skład mieszaniny w odbieralniku posługując się krzywą wzorcową. Wyniki zapisać w tabelce otrzymanej od asystenta. Równanie do obliczania ilości heksanolu Y = 154845x - 7395 y = pole powierzchni piku heksanolu x = ilość heksanolu [ mg ]heksen y* = 175968x* + 1594 y* = pole powierzchni piku heksenu x *= ilość heksenu [ mg]
|
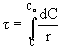 Czas przebywania w reaktorze t
Czas przebywania w reaktorze t