|
WYDZIAŁ CHEMII - ZAKŁAD DYDAKTYCZNY TECHNOLOGII CHEMICZNEJ ADSORPCYJNE OCZYSZCZANIE
GAZÓW
Opracowała dr inż. Jadwiga Skupińska
Emisja par związków organicznych może pochodzić
ze źródeł naturalnych, przemysłowych i wtórnych. Szereg procesów biochemicznych
w otaczającym nas środowisku (np.procesy gnilne) prowadzi do emisji różnorakich
związków organicznych. Jednym z przykładów naturalnego źródła zanieczyszczeń
związkami organicznymi może być dojrzewanie owoców, któremu towarzyszy
wydzielanie się etylenu. Etylen przyspiesza dojrzewanie, stąd w przechowalniach
np.: jabłek konieczne jest usuwanie wydzielającego się etylenu w celu zapobieżenia
przyspieszonemu dojrzewaniu.
Zasady procesu adsorpcji i regeneracji Proces adsorpcji składnika gazowego w pierwszym etapie jest związany z jego wędrówką do powierzchni zewnętrznej adsorbenta. Podobnie jak przy absorpcji etap ten jest określony szybkością dyfuzji cząsteczek zanieczyszczeń w fazie gazowej. Dalszym etapem jest dyfuzja w porach adsorbenta do powierzchni wewnętrznej i adsorpcja na tej powierzchni. Powierzchnia wewnętrzna adsorbenta jest bardzo duża, kilka tysięcy m2/kg. Dyfuzja w porach często jest nazywana dyfuzją wewnętrzną i również może być bardzo wolna i decydować o szybkości całego procesu. Zgodnie z prawami kinetyki szybkość całego procesu jest określana przez jego etap najwolniejszy. Etapy procesu adsorpcji:
Adsorpcja na powierzchni wewnętrznej, adsorbenta, jest etapem szybszym w porównaniu z dwoma poprzednimi. Gdy cząsteczki gazu są adsorbowane (wiązane) na powierzchni stosunkowo małymi siłami, rzędu sił Van der Waalsa, to proces nazywa się adsorpcją fizyczną. Gdy siły wiązania cząsteczek gazu na powierzchni adsorbenta są zbliżone do wiązania chemicznego, proces jest określany jako adsorpcja chemiczna lub chemisorpcja. Wymienione rodzaje adsorpcji zasadniczo różnią się mechanizmem przebiegu procesu. Adsorpcja fizyczna gazu zachodzi z duża szybkością, w niskich temperaturach, w dowolnym miejscu powierzchni adsorbenta Dla przebiegu chemisorpcji w takich warunkach niezbędne jest doprowadzenie tzw. energii aktywacji, koniecznej do wytworzenia odpowiedniego wiązania (kompleksu) adsorbent-substancja adsorbowana. Stwierdzono, że w procesie chemisorpcji tworzy się jednocząsteczkowa warstwa zaadsorbowanych cząsteczek, gdy natomiast podczas adsorpcji fizycznej warstwy te mogą być wielocząsteczkowe. Na odmienność adsorpcji fizycznej i chemisorpcji wskazuje również duża odwracalność adsorpcji fizycznej. Podczas gdy cząsteczki zaadsorbowane fizycznie łatwo jest zdesorbować przez ogrzewaie adsorbenta, to zdesorbowanie cząsteczek związanych chemisorpcyjnie wymaga o wiele większej energii. Ciepło adsorpcji chemicznej jest dużo większe niż adsorpcji fizycznej i jest rzędu ciepła reakcji (20-400).103 kJ/kmol. Dodatkowym procesem, któremu może podlegać gaz podczas adsorpcji jest tzw. kondensacja kapilarna. Przyjmuje się, że adsorpcja w kapilarach (porach) zwilżanych przez ciecz zachodzi przy ciśnieniach niższych od ciśnienia w układzie. Kapilary o najmniejszej średnicy zapełnione zostają w pierwszym rzędzie adsorbowanym składnikiem. Podczas kondensacji kapilarnej jest wydzielane ciepło adsorpcji będące sumą ciepła zmiany fazy z gazowej na ciekłą (ciepło kondensacji) i energii wiązania na powierzchni adsorbenta. Przykładowe wartości całkowitego ciepła adsorpcji dla par substancji organicznych podano w tablicy nr 1. Na zdolność adsorpcyjną adsorbenta wywiera wpływ rozmiar jego ziaren w zakresie od kilku mm do < 20 mikrometrów oraz wielkość porów, których średnica powinna być kilka razy większa od rozmiarów cząsteczek adsorbowanych gazów. Na zdolność danego składnika do adsorpcji wpływa jego masa molowa oraz jego temperatura wrzenia i krytyczna (tabela nr 2). Tablica 1. Całkowite ciepło adsorpcji na
węglu aktywnym.
Tabela 2. Adsorpcja na węglu aktywnym (T
= 288K) w zależności od masy molowej, temperatury wrzenia i temperatury
krytycznej adsorbowanego składnika.
Gdy zaadsorbowana w danych warunkach masa substancji zbliżona jest do ilości równowagowej, następnym etapem procesu adsorpcyjnego oczyszczania gazu jest usunięcie tej substancji z powierzchni adsorbenta, tj. desorpcja, co zwane jest regeneracją adsorbenta. Etapy procesu desorpcji:
Pomimo, że z ekonomicznego punktu widzenia najkorzystniejszą jest wersja z regeneracją adsorbenta i odzyskiem wydzielonej substancji, w praktyce są stosowane również adsorbenty stanowiące ładunek (wsad) jednorazowy. Po wykorzystaniu zdolności sorpcyjnych ładunku stanowi on odpad lub może być spalony, jeżeli sorbentem jest węgiel. Desorpcja może być prowadzona poprzez ogrzewanie adsorbenta, przepływ (przedmuchiwanie) gazu obojętnego przez warstwę nasyconego adsorbenta, oraz przez kombinację wymienionych metod. Ogrzewanie do temperatury wrzenia zaadsorbowanej substancji jest najczęściej stosowaną metodą regeneracji. W przypadku zagrożenia rozkładem substancji desorbowanej stosuje się odpowiednią temperaturę w kombinacji z obniżeniem ciśnienia w układzie. W wyniku desorpcji zaadsorbowanego składnika powietrzem, azotem lub innym gazem obojętnym zanieczyszczenia przechodzą do tego gazu. Ten przypadek regeneracji może być stosowany wówczas, gdy zanieczyszczenia są przeznaczone do spalania. Często stosowaną metodą regeneracji, szczególnie węgli aktywnych, jest działanie parą wodną. Para wodna, adsorbując się, wypiera zaadsorbowane uprzednio substancje, których ciśnienia cząstkowe są mniejsze od równowagowych. Zdesorbowane zanieczyszczenia są kondensowane w nadmiarze pary. Po regeneracji, wodę z adsorbenta usuwa się strumieniem gorącego powietrza. Podczas regeneracji w praktyce nie zachodzi
całkowita desorpcja substancji. Niektóre zanieczyszczenia organiczne, a
szczególnie monomery, mogą podczas regeneracji polimeryzować w porach adsorbenta,
uniemożliwiając powtórne jego stosowanie, up. styren. W wysokich temperaturach
regeneracji niektóre substancje organiczne ulegać mogą rozkładowi (krakingowi),
odkładając się na powierzchniach sorbenta w postaci smoły lub tzw. „nalotów",
co dezaktywuje powierzchnię. Gdy istnieją możliwości wystąpienia takich
przypadków, innymi metodami usuwa się zanieczyszczenia bądź prowadzi desorpcję
w umiarkowanej temperaturze pod obniżonym ciśnieniem działając parą wodną,
lub po wyładowaniu warstwy prowadzi się desorpcję w piecu utleniającym.
W ostatnim przypadku w warunkach kontrolowanych substancje organiczne ulegają
wypaleniu z powierzchni i porów adsorbenta. Po procesie chemisorpcji często
wymagane są specyficzne warunki prowadzenia regeneracji złoża. Desorpcja
wiąże się wówczas zwykle z niszczeniem struktury wiązania adsorbenta.
Adsorbenty 1. Węgiel aktywny Jednym z najstarszych i najpospolitszych
adsorbentów jest węgiel znany jako węgiel aktywowany, węgiel aktywny, węgiel
aktywny-drzewny. Węgiel aktywny otrzymuje się z różnorodnych materiałów
pochodzenia organicznego jak drewno, torf, lignina, pestki owoców, skorupy
orzechów, węgle kopalne i
Ze względu na to, że wytworzone pory są szerokie, materiał ten ma niewielkie właściwości chłonne. Aby uzyskać dobry adsorbent, poddaje się go aktywacji. Proces ten polega zwykle na działaniu na karbonizat parą wodną, dwutlenkiem węgla i tlenem w wysokiej temperaturze, lub prażeniu surowca zmieszanego z czynnikami chemicznymi (chlorek cynku, kwas fosforowy). Proces aktywacji powoduje wytworzenie systemu mikroporów, który nadaje produktowi pożądane właściwości sorpcyjne. Powierzchnia właściwa dobrych węgli aktywnych sięga 1000 m2/g. Węgiel aktywny stosowany w procesach oczyszczania gazów musi mieć postać ziarnistą i odpowiednią wytrzymałość mechaniczną. Węgle aktywne mają budowę mikrokrystaliczną. Ich szkielet zbudowany jest z nieregularnie ułożonych sieci sześcioczłonowych pierścieni węglowych (Rys 1), do których mogą być przyłączone grupy karboksylowe, typu chinonowego i inne. 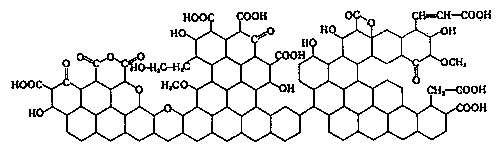 Dobór odpowiedniego surowca, warunków karbonizacji i aktywacji pozwala otrzymać węgle aktywne o bardzo zróżnicowanych właściwościach i różnym przeznaczeniu. W licznych procesach przemysłowych stosowane są lotne rozpuszczalniki, które w trakcie procesu wyparowują w powietrze. Usuniecie par z powietrza zapobiega zanieczyszczaniu atmosfery a jednocześnie pozwala odzyskać rozpuszczalniki, które mogą być powtórnie użyte. Stosuje się w tym celu metodę adsorpcji na węglu aktywnym. Odzyskuje się tą drogą enzymy, benzen, toluen, ksylen, aceton, eter dietylowy, alkohole, chlorowcopochodne węglowodorów, disiarczek węgla i inne rozpuszczalniki. Instalacje do rekuperacji rozpuszczalników składają się zazwyczaj z baterii adsorberów pracujących równolegle. Mieszanina powietrza i par rozpuszczalników po wstępnym odpyleniu transportowana jest do adsorberów za pomocą wentylatorów. Temperaturę gazów utrzymuje się na możliwie niskim poziomie. Nie powinna ona być wyższa niż 40-500C. Gazy o temperaturze wyższej są schładzane w chłodnicach. Instalacja musi pracować tak, by stężenie par substancji organicznej nie przekroczyło polowy granicy wybuchowości, musi tez być wyposażona w system zabezpieczeń przeciwwybuchowych (bezpieczniki ogniowe zapobiegające przenoszeniu się płomienia, urządzenia upustowe odprowadzające gaz na zewnątrz przy nadmiernym wzroście ciśnienia) 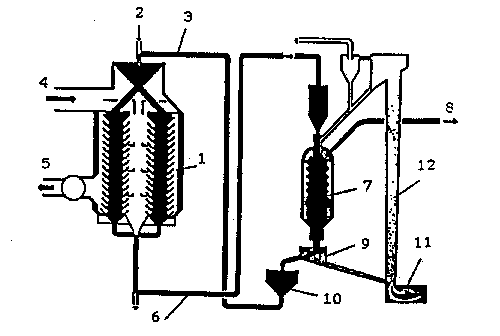 1 - adsorber, 2 - koks aktywny, 3 - koks aktywny z regeneratora, 4 - gaz zanieczyszczony, 5 - gaz oczyszczony, 6 - koks aktywny do regeneracji, 7 - regenerator, 8 - gaz wzbogacony w SO2, 9 - sito wibracyjne, 10 - chłodnica, 11 - komora spalania, 12 - piasek do regeneratora Proces desorpcji przeprowadza się za pomocą
strumienia pary wodnej. Pary wychodzące z adsorbera kierowane są do kondensatora,
gdzie ulegają skropleniu. Dalszy sposób przeróbki kondensatu zależy od
jego składu. Jeżeli zawiera on związki rozpuszczalne w wodzie, to wydziela
się je przez destylację frakcjonowaną w innej instalacji. Jeżeli zdesorbowane
rozpuszczalniki nie mieszają się z wodą, to warstwę organiczną oddziela
się w rozdzielaczu. Złoże węgla po desorpcji suszy się i chłodzi powietrzem
atmosferycznym przed następnym cyklem pracy.
2. Adsorbenty krzemowe Adsorbenty krzemowe stanowią najliczniejszą
klasę sorbentów tlenkowych, takich jak sylikażel, ziemia Fullera, diatomit,
zeolity syntetyczne, sita molekularne. Najbardziej polarne adsorbenty tlenkowe,
np. tlenki glinu, rzadko są stosowane przy adsorpcyjnym oczyszczaniu gazów
odlotowych. Adsorbenty glinowo-krzemowe są stosowane wówczas, gdy ekonomiczna
jest regeneracja ich w wysokich temperaturach, do 700 K. Adsorbentami tymi
są zwykle sita molekularne będące zeolitami syntetycznymi o strukturze,
w której wolne przestrzenie tworzą komory i kanały o ściśle określonych
kształtach i wymiarach . Dzięki temu w danym złożu zeolitu mogą być adsorbowane
cząstki o wymiarach i kształtach dopasowanych do rozmiarów kanałów. Umożliwia
to selektywną sorpcję.
Para wodna adsorbowana jest dobrze na zeolitach
naturalnych krystalicznych lub syntetycznych-hydratach takich metali, jak
wapń, sód, magnez lub ich kombinacje. Budowę zeolitów przedstawiono na
rysunku 3.
Sita molekularne o średnicy porów 0,003 mikrometrów ( A3 ) stosowane są dość powszechnie do usuwania NH3 i pary H2O z gazów obojętnych. Przy średnicy 0,004 mikrometrów ( A4 ) jako typowe adsorbowane są CO2, SO2, H2S, RxS, C2H4. Proste łańcuchy węglowodorów usuwane i rozdzielane są na sitach o wielkości porów 0,005 mikrometrów (A5). Przykładem adsorbentów niekrystalicznych (amorficznych) mogą być sylikażel, tlenek glinu i szereg innych tlenków metali, które zdolne aą do adsorpcji po aktywacji przez dodatek substancji jonowej. Aktywowany tlenek glinu jako wysokoporowaty granulat stosowany jest najczęściej do adsorpcji pary wodnej, po czym jest regenerowany wskutek ogrzewania do temp.460-500 K. Sylikażel jest bezpostaciową uwodnioną krzemionką (SiO2 n H2O) otrzymywaną przez działanie kwasu siarkowego na krzemian sodu (szkło wodne). Rozmiar granul sylikażelu waha się, od ułamka do kilku milimetrów. W zależności od struktury wewnętrznej sylikażel dzieli się na szeroko i wąskoporowaty. Te ostatnie są bardziej wytrzymałe mechanicznie. 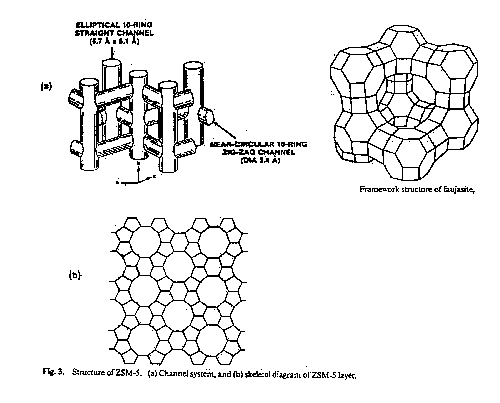 Silikażel jest stosowany przede wszystkim jako środek pochłaniający parę wodną. Przy dużej zawartości pary wodnej w gazie jest stosowany silikażel szeroko porowaty; często stosowany prócz tego w preparatyce katalizatorów jako nośnik. Silikażel nie jest odpowiedni do pracy w wysokich temperaturach. Jego struktura powyżej temp. 530 K ulega degradacji. Silikażel regeneruje się w zakresie temp. 390-470 K. Zdolność sorpcyjną silikażelu w porównaniu z innymi adsorbentami przedstawiono na rys. 4 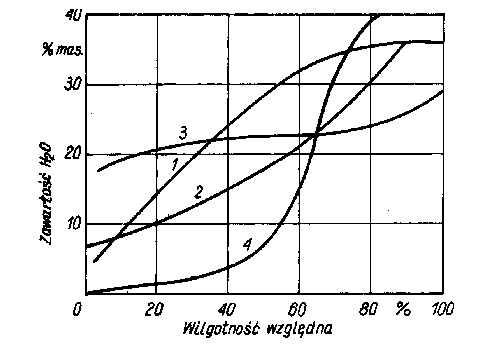 1 - sylikażel, 2 - tlenek
glinu, 3 - sita molekularne, 0,4 mm, 4 - węgiel aktywny
Sorbenty impregnowane dzieli się na trzy grupy, w zależności od funkcji, jaką może spełniać substancja osadzana na ich powierzchni. Należą do nich sorbenty impregnowane reagentem chemicznym, katalizatorem utleniającym lub ułatwlającym rozkład adsorbowanej substancji, katalizatorem działającym okresowo, przyśpieszającym przebieg reakcji pośrednich lub końcowych. Reagenty chemiczne przekształcają substancje zanieczyszczające do postaci nieszkodliwej lub zdolnej do absorpcji. Przykładem może być impregnacja węgla aktywnego solami bromu w procesie adsorpcyjnego oczyszczania gazów z olefin, prowadząca do powstania bromowęglowodorów absorbujących się w wodzie. Sorbenty impregnowane jodem lub siarką stosuje się do usuwania par rtęci, octanem ołowiu do pochłaniania H2S, krzemianem sodu w celu usuwania HF i in. Opracowano również nowe rodzaje adsorbentów zeolitowych do oczyszczania dużych strumieni objętości gazów z lotnych związków organicznych, gdy wymagany jest niski poziom emisji. Produkowane zeolity hydrofobowe mają ściśle określone rozmiary porów, co umożliwia selektywną adsorpcje gazów. Ponadto mają niewielką zdolność adsorpcji par wody i mogą pracować nawet przy 80% wilgotności względnej. Zeolity te są niepalne i ulegają degradacji powyżej 9800C. Stosowane są przede wszystkim tam, gdzie podczas adsorpcji istnieje duże prawdopodobieństwo wybuchu np. par cykloheksanonu lub pożaru. Koszt ich jest jednak wysoki, dlatego używane są tyko tam, gdzie wykluczony jest węgiel. Oprócz prób stosowania włókien wełnianych do usuwania kwaśnych zanieczyszczeń gazowych typu SO2 i NOx opracowano również nową postać adsorbenta węglowego w postaci włókien węglowych, opatentowanego przez firmę japońską Toyobo. Adsorbent stosowany jest w postaci odpowiednio spreparowanych warstw (mat), taśm, arkuszy gładkich lub karbowanych (marszczonych) bądź w postaci .struktury przestrzennej, tzw. plastra miodu. Mikropory na powierzchni włókien mają jednakową strukturę. Struktura porów i ich rozkład zmniejszają znacznie czas adsorpcji i desorpcji, zapewniając ponadto lepszą adsorpcję przy mniejszej masie adsorbenta. Stosowane są do adsorpcji zatężania par rozpuszczalników organicznych z dużych objętości strumieni gazów, 20-100 tys. m3/h, przy małych stężeniach.
Powyższy tekst przygotowano w oparciu o fragmenty książek: 1. J. Warych, „Oczyszczanie przemysłowych
gazów odlotowych”.
Proponowana literatura dla zainteresowanych: 1. Pr. Zbiorowa pod redakcją E.Szczepaniec-Cięciak,
P.Kościelniaka, „Chemia środowiska, ćwiczenia i seminaria”, cz.1, Wyd.
Uniwersytetu Jagielońskiego, Kraków, 1999r.
|